116231
论文已发表
注册即可获取德孚的最新动态
IF 收录期刊
- 3.6 Breast Cancer (Dove Med Press)
- 4.3 Clin Epidemiol
- 2.6 Cancer Manag Res
- 3.2 Infect Drug Resist
- 4.1 Clin Interv Aging
- 6.1 Drug Des Dev Ther
- 4.1 Int J Chronic Obstr
- 8.7 Int J Nanomed
- 2.5 Int J Women's Health
- 3.2 Neuropsych Dis Treat
- 2.4 OncoTargets Ther
- 2.6 Patient Prefer Adher
- 2.6 Ther Clin Risk Manag
- 3.1 J Pain Res
- 3.5 Diabet Metab Synd Ob
- 4.5 Psychol Res Behav Ma
- 3.4 Nat Sci Sleep
- 2.4 Pharmgenomics Pers Med
- 2.6 Risk Manag Healthc Policy
- 4.6 J Inflamm Res
- 2.3 Int J Gen Med
- 3.9 J Hepatocell Carcinoma
- 3.3 J Asthma Allergy
- 2.5 Clin Cosmet Investig Dermatol
- 3.0 J Multidiscip Healthc

通过生物信息学分析鉴定肝癌分子靶基因和关键通路
Authors Zhou L, Du Y, Kong L, Zhang X, Chen Q
Received 10 November 2017
Accepted for publication 16 January 2018
Published 4 April 2018 Volume 2018:11 Pages 1861—1869
DOI https://doi.org/10.2147/OTT.S156737
Checked for plagiarism Yes
Review by Single-blind
Peer reviewers approved by Dr Amy Norman
Peer reviewer comments 3
Editor who approved publication: Dr William Cho
Background and
aim: Hepatocellular carcinoma (HCC) is a major
cause of cancer mortality and is increasing incidence worldwide. The aim of
this study was to identify the key genes and microRNAs in HCC and explore their
potential mechanisms.
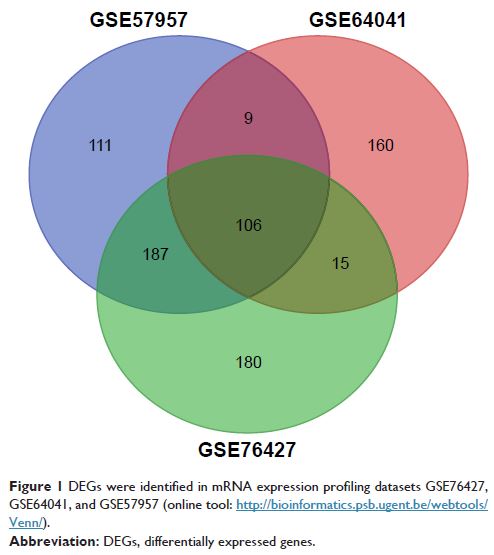
Methods: The gene expression profiles of GSE76427, GSE64041, GSE57957, and
the microRNA dataset GSE67882 were downloaded from the Gene Expression Omnibus
database. The online tool GEO2R was used to obtain differentially expressed
genes (DEGs) and miRNAs (DEMs). The gene ontology and the Kyoto Encyclopedia of
Genes and Genomes pathway enrichment analysis were performed for DEGs using the
Database for Annotation, Visualization, and Integrated Discovery. A
protein–protein interaction (PPI) network of the DEGs was constructed by Search
Tool for the Retrieval of Interacting Genes and visualized by Cytoscape.
Moreover, miRecords was used to predict the target genes of DEMs.
Results: In total, 106 DEGs were screened out in HCC, consisting of 89
upregulated genes and 17 downregulated genes, which were mainly enriched in
biological processes associated with oxidation–reduction process. Besides, the
Kyoto Encyclopedia of Genes and Genomes pathways including chemical
carcinogenesis, drug metabolism-cytochrome P450, tryptophan metabolism, and
retinol metabolism were involved. A PPI network was constructed consisting of
105 nodes and 66 edges. A significant module including nine hub genes, ASPM,
AURKA, CCNB2, CDKN3, MELK, NCAPG, NUSAP1, PRC1, and TOP2A, was detected from
the PPI network by Molecular Complex Detection. The enriched functions were
mainly associated with the mitotic cell cycle process, cell division, and
mitotic cell cycle. In addition, a total of 21 DEMs were identified, including
9 upregulated and 12 downregulated miRNAs. Interestingly, ZBTB41 was the
potential target of seven miRNAs. Finally, the nine hub genes and three
miRNA-target genes expression levels were validated by reverse
transcription-polymerase chain reaction. The relative expression levels of nine
genes (ASPM, AURKA, CDKN3, MELK, NCAPG, PRC1, TOP2A, ZBTB41, and ZNF148) were
significantly upregulated in cancer tissues.
Conclusion: This study identified the key genes and potential molecular mechanisms
underlying the development of HCC, which could provide new insight for HCC
interventional strategies.
Keywords: hepatocellular carcinoma, bioinformatic analysis, differentially
expressed genes, differentially expressed microRNAs