116231
论文已发表
注册即可获取德孚的最新动态
IF 收录期刊
- 3.6 Breast Cancer (Dove Med Press)
- 4.3 Clin Epidemiol
- 2.6 Cancer Manag Res
- 3.2 Infect Drug Resist
- 4.1 Clin Interv Aging
- 6.1 Drug Des Dev Ther
- 4.1 Int J Chronic Obstr
- 8.7 Int J Nanomed
- 2.5 Int J Women's Health
- 3.2 Neuropsych Dis Treat
- 2.4 OncoTargets Ther
- 2.6 Patient Prefer Adher
- 2.6 Ther Clin Risk Manag
- 3.1 J Pain Res
- 3.5 Diabet Metab Synd Ob
- 4.5 Psychol Res Behav Ma
- 3.4 Nat Sci Sleep
- 2.4 Pharmgenomics Pers Med
- 2.6 Risk Manag Healthc Policy
- 4.6 J Inflamm Res
- 2.3 Int J Gen Med
- 3.9 J Hepatocell Carcinoma
- 3.3 J Asthma Allergy
- 2.5 Clin Cosmet Investig Dermatol
- 3.0 J Multidiscip Healthc

已发表论文
综合性的生物信息学分析用于鉴定卵巢癌中的差异表达基因和信号通路
Authors Yang X, Zhu S, Li L, Zhang L, Xian S, Wang Y, Cheng Y
Received 21 September 2017
Accepted for publication 28 November 2017
Published 15 March 2018 Volume 2018:11 Pages 1457—1474
DOI https://doi.org/10.2147/OTT.S152238
Checked for plagiarism Yes
Review by Single-blind
Peer reviewers approved by Dr Akshita Wason
Peer reviewer comments 3
Editor who approved publication: Dr Samir Farghaly
Background: The mortality
rate associated with ovarian cancer ranks the highest among gynecological
malignancies. However, the cause and underlying molecular events of ovarian
cancer are not clear. Here, we applied integrated bioinformatics to identify
key pathogenic genes involved in ovarian cancer and reveal potential molecular
mechanisms.
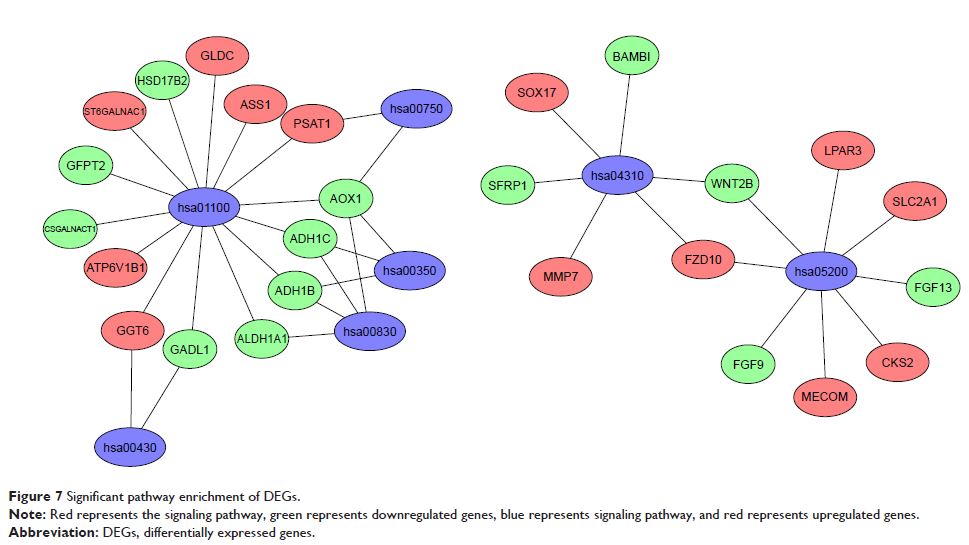
Results: The expression profiles of GDS3592, GSE54388, and GSE66957 were downloaded from the Gene Expression Omnibus (GEO) database, which contained 115 samples, including 85 cases of ovarian cancer samples and 30 cases of normal ovarian samples. The three microarray datasets were integrated to obtain differentially expressed genes (DEGs) and were deeply analyzed by bioinformatics methods. The gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichments of DEGs were performed by DAVID and KOBAS online analyses, respectively. The protein–protein interaction (PPI) networks of the DEGs were constructed from the STRING database. A total of 190 DEGs were identified in the three GEO datasets, of which 99 genes were upregulated and 91 genes were downregulated. GO analysis showed that the biological functions of DEGs focused primarily on regulating cell proliferation, adhesion, and differentiation and intracellular signal cascades. The main cellular components include cell membranes, exosomes, the cytoskeleton, and the extracellular matrix. The molecular functions include growth factor activity, protein kinase regulation, DNA binding, and oxygen transport activity. KEGG pathway analysis showed that these DEGs were mainly involved in the Wnt signaling pathway, amino acid metabolism, and the tumor signaling pathway. The 17 most closely related genes among DEGs were identified from the PPI network.
Conclusion: This study indicates that screening for DEGs and pathways in ovarian cancer using integrated bioinformatics analyses could help us understand the molecular mechanism underlying the development of ovarian cancer, be of clinical significance for the early diagnosis and prevention of ovarian cancer, and provide effective targets for the treatment of ovarian cancer.
Keywords: ovarian cancer, GEO data, integrated bioinformatics, differentially expressed genes
Results: The expression profiles of GDS3592, GSE54388, and GSE66957 were downloaded from the Gene Expression Omnibus (GEO) database, which contained 115 samples, including 85 cases of ovarian cancer samples and 30 cases of normal ovarian samples. The three microarray datasets were integrated to obtain differentially expressed genes (DEGs) and were deeply analyzed by bioinformatics methods. The gene ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichments of DEGs were performed by DAVID and KOBAS online analyses, respectively. The protein–protein interaction (PPI) networks of the DEGs were constructed from the STRING database. A total of 190 DEGs were identified in the three GEO datasets, of which 99 genes were upregulated and 91 genes were downregulated. GO analysis showed that the biological functions of DEGs focused primarily on regulating cell proliferation, adhesion, and differentiation and intracellular signal cascades. The main cellular components include cell membranes, exosomes, the cytoskeleton, and the extracellular matrix. The molecular functions include growth factor activity, protein kinase regulation, DNA binding, and oxygen transport activity. KEGG pathway analysis showed that these DEGs were mainly involved in the Wnt signaling pathway, amino acid metabolism, and the tumor signaling pathway. The 17 most closely related genes among DEGs were identified from the PPI network.
Conclusion: This study indicates that screening for DEGs and pathways in ovarian cancer using integrated bioinformatics analyses could help us understand the molecular mechanism underlying the development of ovarian cancer, be of clinical significance for the early diagnosis and prevention of ovarian cancer, and provide effective targets for the treatment of ovarian cancer.
Keywords: ovarian cancer, GEO data, integrated bioinformatics, differentially expressed genes