116231
论文已发表
注册即可获取德孚的最新动态
IF 收录期刊
- 3.6 Breast Cancer (Dove Med Press)
- 4.3 Clin Epidemiol
- 2.6 Cancer Manag Res
- 3.2 Infect Drug Resist
- 4.1 Clin Interv Aging
- 6.1 Drug Des Dev Ther
- 4.1 Int J Chronic Obstr
- 8.7 Int J Nanomed
- 2.5 Int J Women's Health
- 3.2 Neuropsych Dis Treat
- 2.4 OncoTargets Ther
- 2.6 Patient Prefer Adher
- 2.6 Ther Clin Risk Manag
- 3.1 J Pain Res
- 3.5 Diabet Metab Synd Ob
- 4.5 Psychol Res Behav Ma
- 3.4 Nat Sci Sleep
- 2.4 Pharmgenomics Pers Med
- 2.6 Risk Manag Healthc Policy
- 4.6 J Inflamm Res
- 2.3 Int J Gen Med
- 3.9 J Hepatocell Carcinoma
- 3.3 J Asthma Allergy
- 2.5 Clin Cosmet Investig Dermatol
- 3.0 J Multidiscip Healthc

利用生物信息学方法鉴定黑素瘤中发生表观遗传学改变的基因和潜在的基因靶点
Authors Duan HH, Jiang K, Wei DK, Zhang LJ, Cheng DL, Lv M, Xu YB, He AM
Received 18 July 2017
Accepted for publication 4 October 2017
Published 20 December 2017 Volume 2018:11 Pages 9—15
DOI https://doi.org/10.2147/OTT.S146663
Checked for plagiarism Yes
Review by Single-blind
Peer reviewers approved by Dr Manfred Beleut
Peer reviewer comments 3
Editor who approved publication: Dr Jianmin Xu
Abstract: This study aimed to analyze epigenetically and genetically altered genes
in melanoma to get a better understanding of the molecular circuitry of
melanoma and identify potential gene targets for the treatment of melanoma. The
microarray data of GSE31879, including mRNA expression profiles (seven melanoma
and four melanocyte samples) and DNA methylation profiles (seven melanoma and
five melanocyte samples), were downloaded from the Gene Expression Omnibus
database. Differentially expressed genes (DEGs) and differentially methylated
positions (DMPs) were screened using the linear models for microarray data
(limma) package in melanoma compared with melanocyte samples. Gene ontology
(GO) and pathway enrichment analysis of the DEGs were carried out using the
Database for Annotation, Visualization, and Integrated Discovery. Moreover,
differentially methylated genes (DMGs) were identified, and a transcriptional
regulatory network was constructed using the University of California Santa
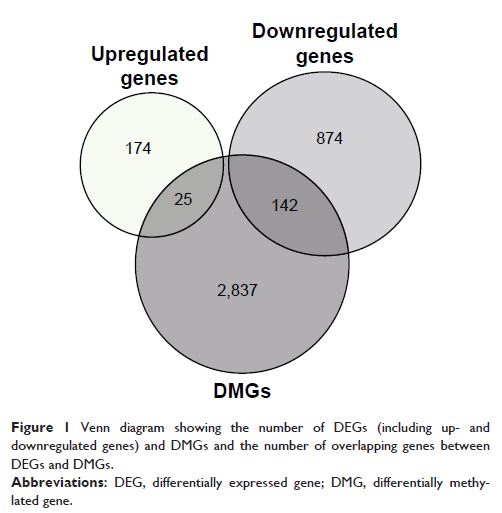
Cruz genome browser database. A total of 1,215 DEGs (199 upregulated and 1,016
downregulated) and 14,094 DMPs (10,450 upregulated and 3,644 downregulated)
were identified in melanoma compared with melanocyte samples. Additionally, the
upregulated and downregulated DEGs were significantly associated with different
GO terms and pathways, such as pigment cell differentiation, biosynthesis, and
metabolism. Furthermore, the transcriptional regulatory network showed that
DMGs such as Aristaless-related homeobox (ARX ),
damage-specific DNA binding protein 2 (DDB2 ), and myelin
basic protein (MBP ) had higher node degrees. Our
results showed that several methylated genes (ARX , DDB2 , and MBP ) may be involved in melanoma
progression.
Keywords: melanoma, DNA
methylation, differentially expressed genes, gene ontology, pathway enrichment
analysis, transcriptional regulatory network